NOTICE
2020 Aug 05: MOCAT is no longer being actively developed and supportedPlease consider switching to newer tools such as NGLess and NGLess meta profilers in conjunction with the mOTUs profiler, the GMGC gene catalog and the EggNOG database and mapper.
Process & analyze metagenomes
MOCAT2 (metagenomic analysis toolkit) is a package for analyzing metagenomics datasets. Currently MOCAT2 supports Illumina single- and paired-end reads in raw FastQ format. Using MOCAT2 you can generate taxonomic and functional profiles, as well as assemblereads and predict genes in assembled sequences. Visit the about or cite us page to see which software is used by MOCAT2.
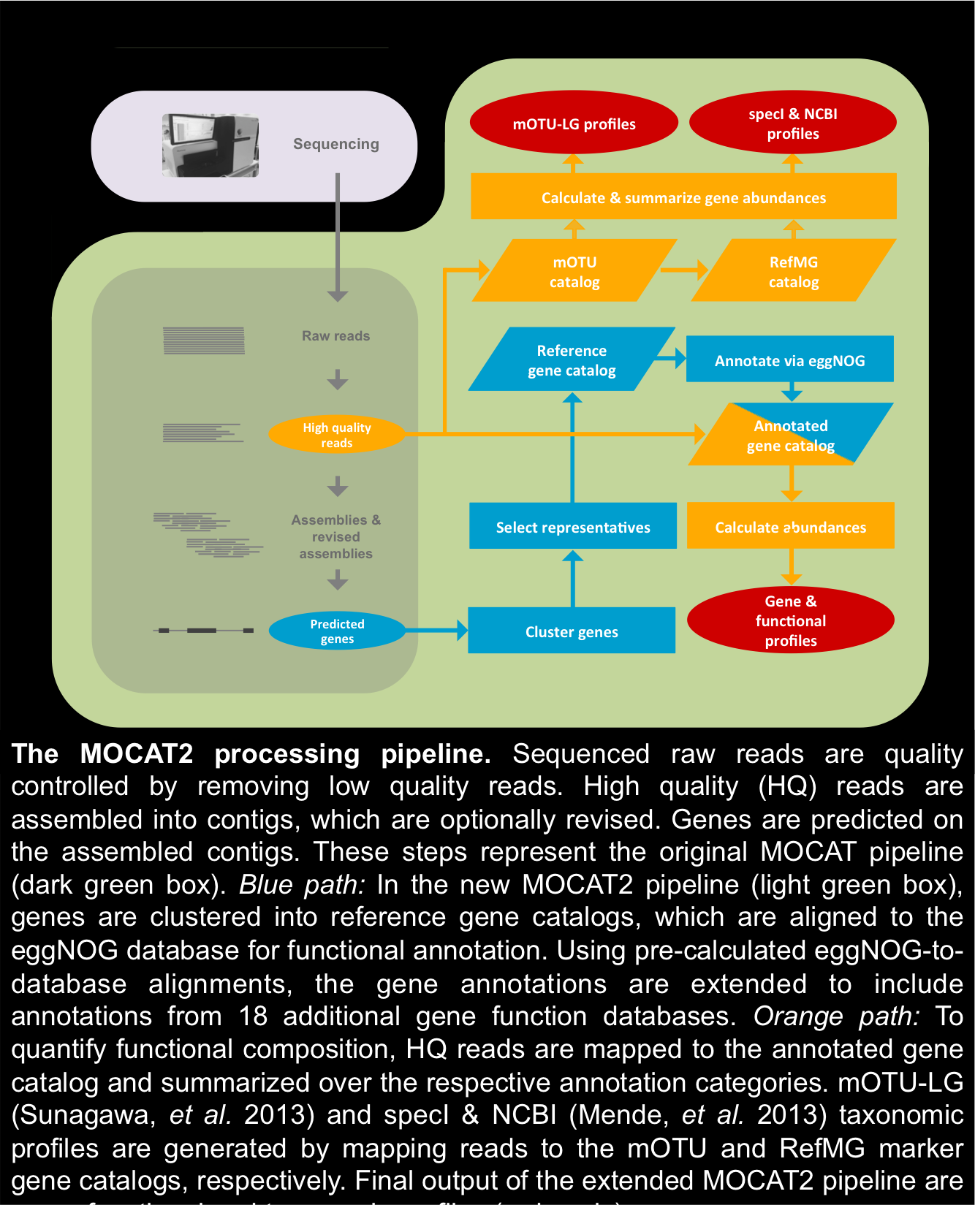
The MOCAT2 pipeline processes raw output files in FastQ format by first quality trimming and filtering the reads.
Optionally it's possible to screen
the reads for contaminants,
if for example you're analyzing
bacterial sequences you can
easily remove any eukaryotic
reads.
It is possible to estimate taxonomic abundances using reference databases. Provided
with MOCAT2 are the RefMG.v1 database (which contains single copy marker genes from 1,753 bacterial reference genomes) and the
mOTU.v1 database (extracted marker genes from 263 metagenomes and 3,496 bacterial genomes). Either of these
databases can be used to generate taxonomic profiles of metagenomes, the first using bacterial references and the
second using single copy marker genes.
Functional profiles can be generated by mapping reads to reference gene catalogs. MOCAT2 comes with annotated reference gene catalogs specific for analyzing bacteria in the human gut, human skin, mouse gut and the oceans. It is also possible to generate your own gene catalog using MOCAT2. The gene catalogs are annotated to 19 gene function databases.
MOCAT supports the assembly of
reads into
scaftigs. After assembly, an
additional assembly
revision step is
performed, to improve the
initial assembly.
After assembly and/or assembly revision, genes can be predicted on the assembled sequences. And then single copy marker genes can be extracted from among the predicted genes.
Predicted genes can be clustered into reference gene catalog. These catalogs can subsequently be annotated to 19 gene funciton databases by aligning reads to the pre-annotated eggNOG database.
